






Washington University Specialized Program of Research Excellence (SPORE) in Leukemia
Washington University
Principal Investigator(s):

Daniel Link, MD
- Principal Investigator(s) Contact Information
- Overview
- Project 1: Chimeric antigen receptor T-cell therapy for T cell malignancies
- Project 2. Memory-like NK cell therapy for AML relapsed after allogenic transplant
- Project 3. Targeted therapy for splicing factor-mutant myeloid malignancies
- Project 4. Targeting ATR in TP53-mutated MDS/AML
- Biospecimen Processing Core
- Bioinformatics and Biostatistics
- Administrative Core
- Developmental Research Program
- Career Enhancement Program
- Institutional SPORE Website
Principal Investigator(s) Contact Information
Daniel Link, MD
Alan and Edith Wolff Endowed Professor;
Chief, Division of Oncology;
Deputy Director, Alvin J. Siteman Cancer Center
660 South Euclid Avenue, CB 8007-29, St.Louis, 63110
(314) 273-6790
Overview
The Washington University SPORE in Leukemia is a highly dynamic translational cancer research program that focuses specifically on leukemias and myelodysplastic syndromes (MDS). We have assembled an outstanding group of investigators with complementary expertise in basic and clinical leukemia research. In this SPORE, we leverage expertise in cancer genomics, immunology, and hematopoiesis to develop innovative translational research in leukemia. Our long-term goal is to develop novel biomarkers and treatments for leukemias and myelodysplastic syndromes and to develop and promote innovative translational leukemia research. To achieve these goals, the following specific aims are proposed.
Aim 1. We will exploit institutional expertise in cancer genomics, immunology, and hematopoiesis to develop novel biomarkers and treatments for leukemias and myelodysplastic syndromes. Basic research at WUSM has led to the development of translational research projects, three of which feature innovative investigator-initiated therapeutic trials for leukemias or MDS (see figure)
Aim 2. We will enhance the infrastructure that supports translational leukemia research. This SPORE will support the following Shared Research Resources: 1) Core A. Biospecimen Processing; 2) Core B. Biostatistics and Bioinformatics; and 3) Core C. Administration.
Aim 3. We will recruit and train new investigators in translational research. This SPORE will support a Career Enhancement Program (CEP) to recruit and mentor new investigators in translational leukemia research, with an emphasis on under-represented in medicine (URiM) investigators. The SPORE has established a successful URiM post-baccalaureate training program. The SPORE also will support a Developmental Research Program (DRP) to promote innovative translational concepts.
Aim 4. We will facilitate inter-SPORE collaboration. Two of the SPORE projects include multi-institutional clinical trials, including participation by peer Leukemia SPORE institutions. We have established a CEP educational exchange and grant review programs with peer Leukemia SPORE institutions. We will continue to organize and participate in joint meetings of with peer Leukemia SPORE institutions.
Washington University SPORE in Leukemia
Project 1. Chimeric antigen receptor T-cell therapy for T cell malignancies
Project Co-Leaders:
John F. DiPersio, MD, PhD (WUSM) (Basic Science Leader)
Mike Rettig, PhD (WUSM) (Basic Science Co-Leader)
Armin Ghobadi, MD (WUSM) (Clinical Co-Leader)
Adult patients with T cell acute lymphoblastic leukemia (T-ALL) exhibit long-term event-free survival of ~40%. Patients with r/r T-ALL have approximate one year and five-year overall survival rates of only 25% and 11%, respectively. Thus, a significant unmet medical need exists for the treatment of r/r T-ALL. We were the first to develop a novel fratricide resistant ‘off-the-shelf’ universal allogeneic CART that targets CD7+ T cell malignancies (UCART7).
Aim 1. We will perform a phase 1 trial evaluating anti-CD7 allogeneic CAR-T (UCART7) in r/r T-ALL (NCT04984356). This first-in-man trial of a universal allogeneic CART to CD7 will consist of a dose escalation phase to determine safety, tolerability, and the recommended phase 2 dose (RP2D) and then an expansion phase at the RP2D to assess efficacy. These initial clinical data will be used to design a future registration study with the hope of getting FDA approval of UCART7 as the first allogenic CART for the treatment of r/r T-ALL. Samples will be obtained to correlate biomarkers (serum cytokines, flow cytometry and expression of specific genes in UCART) with toxicity, efficacy and expansion of UCART7.
Aim 2. We will attempt to enhance the efficacy of UCART7 using a series of novel interventions. These include assessing the role of long-acting cytokines such as NT-I7, N-803, membrane bound IL-15 and membrane bound IL-7. Since CART cell therapy is also associated with life threatening toxicities such a cytokine release syndrome (CRS) we will develop in vitro and preclinical models of CRS and use specific kinase inhibitors to inhibit CRS without limiting CART function. Finally, in order to address UCART7 resistance due to CD7 antigen loss we will develop and fratricide-resistant CD2 and CD7 dual-targeted UCART (UCART2/7).
This project will further the development of a viable ‘off-the-shelf’ therapy for the treatment of T-ALL and potentially other malignancies expressing CD7 or CD2. Additionally, the work proposed here may provide a strategy to treat relapse after UCART7 and prevent life-threatening CRS.
Project 2. Memory-like NK cell therapy for AML relapsed after allogenic transplant
Project Co-Leaders:
Todd Fehniger, MD, PhD (WashU)
Amanda Cashen, MD (WashU)
Immunotherapy is a type of cancer treatment that alters components of the immune system to increase their response to cancer. This project seeks to develop new forms of immunotherapy for a difficult-to-treat blood cancer, acute myeloid leukemia (AML). For patients with AML, standard treatments include chemotherapy, and if there is a higher risk of relapse, hematopoietic stem cell transplant (HCT). If AML relapses after HCT there are few standard treatment options that work, and hence, a great need to develop new treatments.
This project addresses this unmet clinical need by using an immune cell, the natural killer (NK) cell, to treat AML. In a clinical trial, patients who have AML that relapsed after HCT receive ‘super charged’ memory-like NK cells that have been programmed via protein hormones (cytokines) for improved anti-tumor attack. NK cells collected from the original HCT donor will be used to generate the therapeutic memory-like NK cells, which are activated by interleukins 12, 15, and 18 overnight, prior to infusion into the patient. Patients first receive mild chemotherapy to make space within the patient for the cells to expand, persist, and fight the leukemia. As part of this study, the patients’ samples will be examined for the quality and quantity of the transferred NK cells. Leukemia cells will be tested for properties that result in resistance to NK cells.
In a second part of the project, a pre-clinical study will test recently identified approaches to further improve memory-like NK cell responses to cancer. These include blocking an inhibitory or “off” signal to the NK cell delivered by the cancer cells, as well as engineering memory-like NK cells to express chimeric antigen receptors that help them “see” and therefore attack cancer cells more effectively. This part of the project will lead to new clinical trial ideas to test improved versions of memory-like NK cells in combination with NK cell-checkpoint blockade.
Collectively, this project will test a promising cellular therapy for leukemia in a clinical trial, and develop new ways to further increase the activity of NK cells against cancers.
Project 3. Targeted therapy for splicing factor-mutant myeloid malignancies
Project Co-Leaders:
Matthew Walter, MD (Wash U) (Basic Science Co-Leader)
Timothy Graubert, MD (MGH) (Clinical Co-Leader)
The long-term goal of this project is to develop novel therapies that exploit vulnerabilities induced by RNA splicing factor gene mutations in patients with myeloid neoplasms, including myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML). Preliminary results from our investigator-initiated clinical trial show that inhibiting ATR, a protein involved in DNA repair, using a single drug induces objective responses with acceptable toxicity in patients, but combination therapy will be required for more durable clinical benefit. This project will optimize the combination of ATR and PARP1 inhibition in preclinical models, providing a rationale for the next phase of clinical investigation.
Aim 1: We will use multiple in vivo preclinical models to test the efficacy and safety of targeting both ATR and PARP1, another protein involved in DNA repair, in myeloid malignancies with splicing factor mutations. We will explore CHK1 inhibition as an alternative strategy to target the ATR signaling pathway.
Aim 2: We will explore mechanisms of sensitivity and resistance to ATR inhibition using clinical samples, clinically relevant genetically-engineered mouse models, and gene-edited human leukemia cells.
The overall translational impact includes:
- providing preclinical proof of concept for dual targeting of ATR and PARP1 in myeloid malignancies with splicing factor mutations;
- developing a preclinical rationale for CHK1 inhibition as an alternative strategy to target ATR signaling;
- identifying biomarkers of sensitivity to ATR pathway inhibition, and
- understanding molecular mechanisms of resistance to ATR inhibition.
Project 4. Targeting ATR in TP53-mutated MDS/AML
Project Co-Leaders:
Geoffrey Uy, MD, PhD; (WashU) (Clinical Co-Leader)
Daniel C. Link, MD; (WashU) (Basic Science Co-Leader)
Mutations of TP53 are found in a significant percentage of cases of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). These mutations have a negative impact on prognosis, and standard therapies have limited effectiveness. Hypomethylating agents have shown some therapeutic activity, but complete mutation clearance is rare, leading to relapse. Our research suggests that TP53-mutated cells are sensitive to the combination of decitabine (a hypomethylating agent) and an ATR inhibitor (see figure). The study aims to characterize the therapeutic potential of this combination and investigate the underlying molecular mechanisms. In Aim 1, we will perform a clinical trial of an ATR inhibitor in combination with decitabine for patients with AML or MDS carrying TP53 mutations. Aim 2 will determine mechanisms by which this drug combination is selectively active in TP53-mutated AML/MDS. The ultimate goal is to provide evidence of safety, target engagement, and efficacy to support further clinical trials. Additionally, the molecular studies may reveal new insights into the sensitization of AML cells and identify potential new targets for treatment.
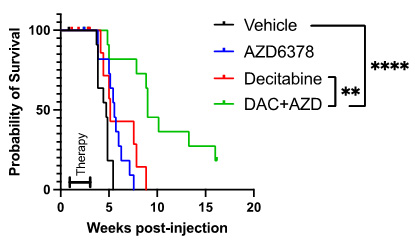
Improved survival of TP53-mutated AML in mice treated with decitabine (DAC) plus ATR inhibitor (ATRi) compared with decitabine alone.
Biospecimen Processing Core (Core A)
Core Directors
Meagan A. Jacoby, MD, PhD (WashU)
Mark A. Watson, MD, PhD (WashU)
The Biospecimen Processing Core (Core A) provides high-quality, clinically annotated biospecimens to all SPORE research projects and serves as a critical biospecimen resource to the broader leukemia research community. This core is responsible for identifying and enrolling every consenting patient referred to the Siteman Cancer Center with a newly diagnosed hematologic malignancy (excluding multiple myeloma). Biospecimens from these patients are collected at multiple timepoints, processed, cryopreserved, and stored for future studies. Detailed clinical data associated with each patient and biospecimen collection timepoint are collected prospectively to facilitate future sample selection and correlative analyses. This Core expands upon an existing CAP-accredited SCC biobank with additional, focused expertise, effort, and enabling technologies to comprehensively collect, annotate, process, quality review, and distribute biospecimens specifically for translational leukemia and leukemia-related cancer research. A further description of biospecimen resources available to the broader research community can be found on the NCI Specimen Resource Locator.
Bioinformatics and Biostatistics (Core B)
Core Directors:
Graham A. Colditz, MD, DRPh (Wash U)
Malachi Griffith, PhD (Wash U)
The Bioinformatics and Biostatistics Core (Core B) will support four leukemia research projects within this SPORE. The primary aim of Core B is to provide robust bioinformatics and statistical support to ensure rigorous and reproducible multi-omics analyses across preclinical studies, clinical trials, and clinical correlative studies. Core B’s services include guiding experimental design, data processing, and comprehensive analysis for a range of data types, including bulk RNA-seq, single-cell RNA-seq, CITE-seq, TCR-seq, whole genome sequencing (WGS), whole genome bisulfite sequencing (WGBS), ATAC-seq, scATAC-seq, CRISPR/Cas9 screens, and CyTOF experiments. By leveraging advanced bioinformatics tools and statistical methodologies, Core B enhances the precision and depth of data interpretation. In supporting the projects, Core B will enhance understanding of new treatment avenues pursued by each project including chimeric antigen receptor T-cell (CART) therapy for T-cell cancers, natural killer (NK) cell therapy for acute myeloid leukemia (AML), and targeted therapies for specific gene mutations in myeloid cancers. Through rigorous design and analysis of clinical trials and preclinical studies, Core B ensures that each project achieves its scientific objectives. Core B fosters a collaborative environment, offering consultation and training to researchers, thereby promoting the effective application of bioinformatics and biostatistics in leukemia research.
Administrative Core (Core C)
Core Director:
Daniel C. Link, MD (WashU)
The Administration Core will provide executive oversight and administrative support for all of the projects and cores that comprise the Leukemia SPORE. The goal of the Administration Core is to monitor the activities of all of the program components, comply with all local and federal guidelines for grant administration, and facilitate communication and collaboration among the program members and with other Leukemia SPOREs. Accordingly, the specific aims of the Administration Core are as follows:
Aim 1. To facilitate intra- and inter-SPORE communication and collaboration.
Aim 2. To provide administrative and fiscal oversight and support for all SPORE components.
Aim 3. To coordinate activities of the SPORE Developmental Research Program.
Aim 4. To coordinate activities of the SPORE Career Enhancement Program.
Aim 5. To assist investigators with the preparation of scholarly presentations, publications, regulatory documents, and all other SPORE-related paperwork.
Aim 6. To encourage diversity and equity in SPORE activities.
Aim 7. To ensure that advocacy issues are addressed and included in all aspects of research involving patients.
Washington University SPORE in Leukemia Organization
Developmental Research Program
Core Directors:
Laura Schuettpelz, Md, PhD (WashU)
Daniel C. Link, MD; (WashU)
The overall goal of the Leukemia SPORE Developmental Research Program (DRP) is to identify and support developmental research projects in leukemia for future peer-reviewed funding and/or future independent SPORE projects. Projects supported under the DRP will expand the scope of translational research and increase the number of investigators committed to leukemia research. The DRP will work in tandem with the Career Enhancement Program (CEP) to assist in the development of junior investigators and in the recruitment and mentoring of Underrepresented in Medicine (URiM) investigators. To accomplish these goals, the DRP aims to: 1) support developmental research projects in leukemia for future incorporation as full SPORE projects or application for other major peer-reviewed funding; 2) foster collaborations between basic and clinical researchers; 3) provide mentoring to junior faculty.
Career Enhancement Program
Program Directors:
Matt Walter, MD (Wash U)
Geoffrey Uy, MD (Wash U)
Timothy J. Ley, MD (Wash U)
The Leukemia SPORE Career Enhancement Program (CEP) has a goal of recruiting and supporting a diverse set of new investigators in the field of translational leukemia research. To accomplish this objective, the CEP will provide financial support and mentored research training. It will leverage institutional strengths to recruit basic scientists and clinical investigators from varied disciplines and backgrounds to promote multidisciplinary translational research. These goals will be accomplished in three ways: 1) We will recruit and financially support new investigators in the field of translational leukemia research. 2) We will provide training and mentoring to junior faculty from all backgrounds in translational leukemia research. CEP will work with new investigators to craft an individualized career development plan that may combine didactic coursework, patient care, and career skills tailored to their individual goals. 3) We will foster inter-SPORE collaborations. We have established educational exchanges to provide CEP awardees the opportunity to present their research and meet with the leadership at a peer Leukemia SPORE Institution.