






SPORE in Breast Cancer
Vanderbilt University Medical Center
Principal Investigators:

Jennifer A. Pietenpol, PhD

Ben Ho Park, MD, PhD
- Principal Investigator(s) Contact Information
- Overview
- Project 1: Mechanisms of Resistance to Endocrine Therapy in ER+ Breast Cancer
- Project 2: Strategies to Improve Outcomes for Triple-Negative Breast Cancer Patients Integrating Subtype-Specific Genomic and Immune-Based Discoveries
- Project 4: Targeting Antigen Presentation to Improve Immunotherapy Responses in Breast Cancer
- Project 5: Liquid Biopsied to Guide Therapies in Triple-Negative Breast
- Supplement Project: Evaluation of Barriers to Completion of Breast Cancer Treatment at a Tertiary Teaching and Referral Center in Kenya — SPORE Supplement
- Administration and Engagement Core
- Biostatistics and Bioinformatics Core
- Pathology and Tissue Informatics Core
- Immunophenotyping Core
- Developmental Research Program
- Career Enhancement Program
- Institutional SPORE Website
Principal Investigators Contact Information
Jennifer A. Pietenpol, PhD
Chief Scientific and Strategy Officer
Executive Vice President for Research
Brock Family Directorship in Career Development
Professor of Biochemistry and Otolaryngology
Vanderbilt University Medical Center
1161 21st Ave. S, D-3300 MCN
Nashville, TN 37232
(615) 936-2033
Ben Ho Park, MD, PhD
Benjamin F. Byrd Jr. Professor of Oncology
Director, Vanderbilt-Ingram Cancer Center
Professor of Medicine Division of Hematology and Oncology
Vanderbilt University Medical Center
2220 Pierce Ave, PRB 698
Nashville, TN 37232
(615) 936-1374
Overview
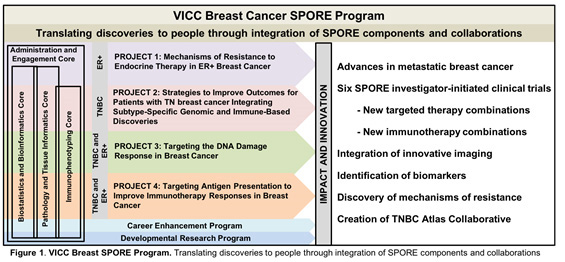
This is the third competing renewal application of the Vanderbilt-Ingram Cancer Center (VICC) SPORE in Breast Cancer, thanks to a very active and strong multidisciplinary VICC Breast Cancer Program that is robustly supported by the SPORE mechanism. Our overall goal remains the same: To conduct collaborative, multidisciplinary and mechanism-based translational research that will have the highest possible impact for women and men with or at risk for breast cancer. After internal and external planning and evaluation, we present four bi-directional translational projects addressing basic and clinical research questions of importance in human breast cancer: Projects 1 and 2 are continuation projects; Projects 4 and 5 are new projects that have evolved from CEP and DRP awards in the current funding cycle. Our new Immunophenotyping Core (IPC), which includes innovative Immuno-PET imaging, along with the highly used Pathology and Tissue Informatics (PTIC), Biostatistics and Bioinformatics (BBC) and Administrative and Engagement Cores (AEC) give key support to SPORE investigators (Fig. 1). Six interventional therapeutic clinical trials are associated with the highly translational projects in this proposal, all based on extensive collaboration between world- class basic and clinical investigators.
Project 1: Mechanisms of Resistance to Endocrine Therapy in ER+ Breast Cancer
Project Co-Leaders:
Brent Rexer, MD, PhD (clinical)
Carlos L. Arteaga, MD (basic)
Specific Aims:
Aim 1: To elucidate the mechanisms by which FGFR1 amplification confers resistance to the combination of fulvestrant and CDK4/6 inhibitors in ER+/FGFR amplified breast cancers
Aim 2: To conduct a randomized phase II trial of the ER downregulator fulvestrant plus palbociclib ± the pan-FGFR inhibitor erdafitinib in patients with ER+/FGFR-amplified metastatic breast cancer
Aim 3: To discover mechanisms of resistance to fulvestrant/palbociclib ± erdafitinib in organoids derived from tumors progressing in the clinical trial proposed in Aim 2
The Project 1 team profiled 155 HR+/HER2- early breast cancers from 143 patients treated with the aromatase inhibitor letrozole for 7-21 days before surgery (Science Translational Medicine 2017 PMC5723145). Whole-exome sequencing (WES) revealed a correlation between 8p11-12 and 11q13 gene amplifications, including FGFR1 and CCND1, respectively, and high on-treatment Ki67. Of note, FGFR1 is amplified in ~15% of early ER+ breast cancers where it is associated with early relapse following adjuvant tamoxifen therapy and poor patient outcome. Additionally, ctDNA data from patients enrolled in MONALEESA-2, the registration trial of ribociclib, a CDK4/6 inhibitor, showed that patients with FGFR1 amplification in ctDNA exhibited a progression-free survival of 10.6 months vs. 24.8 months in patients with wild-type FGFR1. Subsequent to these findings, Project 1 revealed that co-amplification of FGFR1 and CCND1 were associated with endocrine therapy and CDK4/6 inhibitor resistance, that could be reversed by treatment with FGFR1 and CDK4/6 inhibitors (Clin Cancer Res, 2017 PMID 29284706), and reported marked HR+/FGFR1-amplified patient-derived xenografts tumor regressions with fulvestrant, palbociclib (a CDK4/6 inhibitor) and erdafitinib (a pan-FGFR tyrosine kinase inhibitor) (Nat Commun, 2019 PMID 30914635). Based on the above preclinical data, in 2016, Dr. Mayer successfully competed for funding of a three-year Pfizer ASPIRE Breast Cancer Research Award entitled “Phase Ib trial of fulvestrant, palbociclib and erdafitinib in HR+/HER2-/FGFR-amplified metastatic breast cancer,” which now funds one of the highly translational, multi-institutional trials that will test the hypothesis that FGFR inhibitors should be tested in combination with ER antagonists and CDK4/6 inhibitors in patients with HR+/FGFR amplified breast cancer.
Project 2: Strategies to Improve Outcomes for Triple-Negative Breast Cancer Patients Integrating Subtype-Specific Genomic and Immune-Based Discoveries
Project Co-Leaders:
Vandana G. Abramson, MD (clinical)
Jennifer A. Pietenpol, PhD (basic)
Specific Aims:
Aim 1: To determine mechanisms by which standard chemotherapies modulate tumor immune response in combination with checkpoint immunotherapy
Aim 2: To determine mechanisms of immune-modulation in metastatic TNBC subtypes
Aim 3: To evaluate the feasibility of targeting the tumor microenvironment myeloid-derived cells
The Project 2 team is using the discovery framework of preclinical models, genomic tools and TNBC patient tumor biospecimens to achieve improved outcomes for TNBC patients through use of subtype-informed combination immunotherapy. In the prior trial “Randomized phase II neoadjuvant study of cisplatin and paclitaxel with or without everolimus in patients with TNBC” (NCT00930930; Clin Cancer Res, 2017 PMC5540799), they revealed that cisplatin caused increases in TILs in M subtype tumors, as 40.6% of tumors treated with cisplatin had increased PD-L1 expression. The increased PD-L1 expression provided the rationale for the initiation of a phase II clinical trial (NCT03206203) investigating the combination of carboplatin with the anti-PD-L1 antibody atezolizumab with intent to identify biomarkers of response, led by Dr. Abramson (BC). The collection and comprehensive analysis of the biospecimens relative to clinical response enables this project to test the hypothesis that patients receiving carboplatin in combination with atezolizumab will have a better response to PD-L1 treatment. If proven, would suggest carboplatin treatment inhibits MDSC- and Treg-mediated suppression of TIL infiltration.
Project 4: Targeting Antigen Presentation to Improve Immunotherapy Responses in Breast Cancer
Project Co-Leaders:
Laura Kennedy, MD, PhD (clinical)
Justin M. Balko, PharmD, PhD (basic)
Specific Aims:
Aim 1: Determine if MEKi-mediated antigen presentation upregulation predicts immunotherapy outcome in breast cancer patient tumors
Aim 2: Determine the mechanism whereby MEK suppresses anti-tumor immunity
The Project 4 team showed that MEK activation suppresses tumor cell surface MHC class I and II and PD-L1 expression in breast cancer. Conversely, inhibition of MEK enhanced these factors, blocked naïve CD8+ T cell priming, and protected CD8+TIL cytotoxicity by preventing apoptosis driven by chronic TCR stimulation (Clin Cancer Res, 2015 PMC4794351). In luminal-like breast cancer models (MMTV-neu and MMTV-PyVmT), a combination of MEK inhibition and PD-1/PD-L1 inhibitors induced profound anti-tumor effects . Based on these preliminary data, in 2016, Drs. Mayer and Hope Rugo, MD (UCSF) successfully competed for funding of a three-year Breast Cancer Research Fund (BCRF)-Pfizer award entitled “Immunotherapy combination strategies to treat TNBC: a multicenter, multi-arm Translational Breast Cancer Research Consortium (TBCRC) study”. This is a multicenter, open-label, four-arm phase II trial that will evaluate the anti-tumor effect of a PD-L1 inhibitor alone or in combination with either a MEK inhibitor, an OX40 agonist or a 41BB agonist in patients with metastatic TNBC. In 2017, Drs. Mayer and Balko were awarded a three-year Stand Up to Cancer (SU2C)-American Association for Cancer Research (AACR) Catalyst Research Grant titled “Immunotherapy Combination Strategies in ER+ Metastatic Breast Cancer.” This is a multicenter, open-label, two-arm phase Ib/II trial that will evaluate the safety and anti-tumor effect of a PD-L1 inhibitor in combination with a MEK inhibitor in patients with endocrine therapy-refractory TP53-mutated ER+ metastatic BC, and a PD-L1 inhibitor in combination with an MDM2 antagonist in patients with endocrine therapy-refractory TP53-wildtype ER+ metastatic BC (VICCBRE17017/ NCT03566485). These trials will test for synergy between MEK inhibition and anti-PD-L1 therapy, identify predictive biomarkers, as well as validate the clinical utility of new therapeutic combinations that promote anti-tumor immunity through enhancement of antigen presentation.
Project 5: Liquid Biopsied to Guide Therapies in Triple-Negative Breast
Project Co-Leaders:
Vandana G. Abramson, MD (clinical)
Ben Ho Park, MD, PhD (basic)
Specific Aims:
Aim 1: To determine whether patients with metastatic TNBC who undergo treatment changes guided by ctDNA dynamics demonstrate improved PFS compared to control patients assessed conventionally with imaging alone
Aim 2: To validate and functionally assess CHIP mutations as a predictive/prognostic marker for poor response to therapies and worse PFS/OS in patients with metastatic TNBC
Project 5 will evaluate the clinical utility of using cell-free DNA (cfDNA) in metastatic triple-negative breast cancer (TNBC) patients to guide clinical decision-making. In aim 1, Project 5 will conduct a randomized control trial to determine if instituting second line therapy with sacituzumab earlier than the first restaging scan can afford better outcomes in TNBC. If it is determined first line chemotherapy is ineffective by measuring quantitative levels of circulating tumor DNA (ctDNA) before and after the initial chemotherapy dose, sacituzumab will be introduced earlier. In aim 2, Project 5 will determine whether clonal hematopoiesis of indeterminant potential (CHIP) mutations found in cfDNA correlate with worse outcomes in patients on our study. Blood cells with and without CHIP mutations will be analyzed from patient samples and assessed on their effects within different subtypes of TNBC cell lines using ex vivo assays. The results of this aim will identify new potential targets (CHIP clones) that could positively affect outcomes for patients with metastatic TNBC.
Supplement Project: Evaluation of Barriers to Completion of Breast Cancer Treatment at a Tertiary Teaching and Referral Center in Kenya — SPORE Supplement
Supplement Project Co-Leaders
Rondi Kauffmann, MD, MPH, FACS (Vanderbilt University Medical Center)
Beryl Akinyi Ooro, MBChB (AIC Kijabe Hospital, Kenya)
Helena Musau, MBChB (AIC Kijabe Hospital, Kenya)
There is a growing body of literature highlighting the dearth of education in the conduct of research on the African continent. There is need for structured mentorship to train clinician-scientists in Africa to conduct high-quality research that addresses community-specific disease burden. In this research program that is a supplement to the Vanderbilt-Ingram Cancer Center (VICC) Breast SPORE, we will leverage the existing partnership between the Vanderbilt University Medical Center and AIC Kijabe Hospital in Kenya, to further build upon a longitudinal, bi-directional global breast cancer research program.
In Kenya, breast cancer is the most common cancer in women, and has the third highest mortality. Unfortunately, a large percentage of women present with metastatic disease, and even those presenting at an earlier stage often face significant delays in management at multiple stages of the disease continuum. The underlying causes are multi-factorial and include low levels of education about breast disease, long distances to healthcare facilities, high levels of poverty, and scarcity of healthcare personnel and treatment facilities. This study aims to address the lack of understanding of breast cancer care in Kenya in order to identify strategies to tackle barriers to care and improve breast cancer survival. To address this gap, a multi-disciplinary study is being conducted by two Kenyan clinician-scientists in partnership with VICC Breast SPORE investigators.
Specific Aims:
Aim 1: To elucidate the socio-demographics, clinical characteristics, tumor characteristics, treatment and survival outcome for patients diagnosed with breast cancer in Kijabe, Kenya between 2010-2021
Aim 2: To prospectively follow patients diagnosed with breast cancer at Kijabe Hospital through their continuum of breast cancer to identify the factors and system dynamics associated with treatment default or completion.
Aim 3: To assess the feasibility of the use of sentinel node biopsy to stage the axilla in early-stage breast cancer in Kenya.
Administration and Engagement Core
Core Directors:
Ben Ho Park, MD, PhD
Jennifer Pietenpol, PhD
Specific objectives: Management of SPORE resources, provision of quality control, and active communication and engagement, including fostering interaction amongst SPORE components and collaborators, other SPOREs, the patient and advocate community and the National Cancer Institute (NCI).
Biostatistics and Bioinformatics Core
Core Director:
Yu Shyr, PhD
Specific objectives: Development of experimental designs, power analysis and sample size estimation, data quality control, statistical/bioinformatics analysis and interpretation of findings, and collaboration on presentation of results.
Pathology and Tissue Informatics Core
Core Directors:
Travis Osterman, DO, MS
Melinda Sanders, MD
Specific objectives: Clinical trial-related tissue collection, procurement for the Vanderbilt Breast Tissue and Body Fluids Repository, provision of specialized technical and diagnostic histopathology services, informatics support (de-identification, histopathological and clinical annotation, tracking and distribution of breast tissues) for translational research by SPORE investigators, linking specimen-specific information to immunohistochemistry (IHC), and Next Generation Sequencing (NGS) data permitting queries to inform future investigations. Pathology assessments will be made by research and licensed clinical pathologists of the PTIC for clinical/correlative studies within all SPORE clinical trials. The PTIC will also manage the Breast SPORE TNBC Collaborative Atlas.
Immunophenotyping Core
Core Directors:
Kimberly Dahlman, PhD
Jeffrey Rathmell, PhD
Specific objectives: Provide state-of-the-art comprehensive assays that range from validated single cell sequencing of T cells for both α and β chain of TCR and validated Mass Cytometry by Time of Flight (CyTOF) assays; provide established immunological assays — ELISPOT, immunohistochemistry/immunofluorescence with digital quantitation, flow cytometry, Luminex-based cytokine profiling assays, and NGS; provide both immune-cellular phenotyping and immune-genomic phenotyping; support murine model preclinical studies that require immune-cellular phenotyping.
Developmental Research Program
Program Directors:
Scott Hiebert, PhD
Ben Ho Park, MD, PhD
Jennifer A. Pietenpol, PhD
During the current project period, the SPORE Developmental Research Program awarded 15 pilot projects to 14 investigators, of which four were junior faculty at the time of award. All are from nine primary departments in the School of Medicine (Department of Medicine, Biochemistry, Biostatistics, Chemistry, Biomedical Informatics, Cell and Developmental Biology; Pathology, Microbiology & Immunology, Pediatrics, Pharmacology) and six of the eight research programs at VICC (Signal Transduction & Chemical Biology, Host-Tumor Interactions, Genome Maintenance, Translational Research and Interventional Oncology, Breast Cancer, and Cancer Health Outcomes and Control). Although eight of the 15 projects were awarded at or after April 2016 and the PI of a ninth project was recruited to the University of Texas Health Science Center, three of the remaining awardees have obtained R01 or R01-equivalent grants for a combined amount of $2.5M (direct costs), $3.6M (total costs). This return in extramural grant dollars is impressive given the $667,149 overall investment made in the DRP. There are at least 27 published manuscripts supported by this research. Finally, all awardees have presented their research at SPORE monthly meetings or the VICC Breast Cancer Program and SPORE Monthly Series.
Career Enhancement Program
Program Directors:
Jin Chen, MD, PhD
Ben Ho Park, MD, PhD
Jennifer A. Pietenpol, PhD
During the current project period, the SPORE CEP supported 11 investigators (13 total awards): three physician-scientists and eight basic scientists from three Departments in the School of Medicine at Vanderbilt (Radiology and Radiological Sciences, Medicine, and Biochemistry), the Department of Biochemistry and Cancer Biology at Meharry Medical College and four research programs at VICC (Genome Maintenance, Translational Research and Interventional Oncology, Host-Tumor Interactions and Breast Cancer). Thirty publications have resulted from CEP awardees. Six of the awardees have obtained extramural grants for a total combined budget of over $6.8M (direct costs), $9.2M (total costs). This return in extramural grant dollars far surpasses the investment made in the CEP. In addition, two awardees are co-leaders in two of the translational projects in the proposed SPORE, two other awardees (Drs. Hanker and Lehmann) are working with project leaders as basic co-investigators and one awardee (Dr. Houghton) is working with core directors as a co-investigator.