






Developmental and Hyperactive Ras Tumor (DHART) SPORE
Trustees of Indiana University and The Regents of the University of California, San Francisco
Principal Investigator(s):

D. Wade Clapp, MD

Kevin Shannon, MD
- Principal Investigator(s) Contact Information
- Overview
- Project 1: From neurofibroma to MPNST: models, biology and translation to clinic
- Project 2: NF1-associated glioblastoma
- Project 3: A high content clinical trial of the MEK inhibitor Trametinib in JMML
- Administrative Core
- Omics Core
- Biospecimen and Pathology Core
- Developmental Research Program
- Career Enhancement Program
- Institutional SPORE Website
Principal Investigator(s) Contact Information
D. Wade Clapp, MD
Chairman, Department of Pediatrics
Richard L. Schreiner Distinguished Professor of Pediatrics, Microbiology & Immunology,
Biochemistry & Molecular Biology, and Medical and Molecular Genetics
Indiana University School of Medicine
1130 W Michigan St, Fesler Hall 320
Indianapolis, IN 46202
(317) 278-5809
Kevin Shannon, MD
American Cancer Society Research Professor
Auerback Distinguished Professor of Molecular Oncology
University of California, San Francisco, Mission Bay Campus
1450 3rd Street; Room 263
San Francisco, CA 94158-9001
(415) 476-7932
Overview
The overall goal of this Developmental and Hyperactive Ras Tumor (DHART) SPORE is to address this unmet medical need by performing integrated, mechanistically based translational research that will lead to effective targeted molecular therapies for neoplasms and cancers characterized by Neurofibromatosis type 1 (NF1) mutations. Our program is comprised of a highly integrated group of translational scientists that are addressing central issues for rare tumors driven by hyperactive Ras signaling in the pediatric, adolescent, and young adult (AYA) population. The research included in this program involves long-standing collaborative relationships of multiple experts with diverse scientific capabilities. Our multi-disciplinary group is working to deploy novel organizing principles and approaches to address the key challenge of how to accelerate new therapies for uncommon (“orphan”) benign and malignant tumors across an organ spectrum that is characterized by mutations in the NF1 tumor suppressor gene. NF1 is the most common inherited cancer predisposition syndrome, and persons with NF1 have a markedly increased incidence of developing specific tumors.
This program encompasses three highly integrated projects and three cores that seek to develop more effective and less toxic therapies for three aggressive cancers — MPNST, GBM, and JMML (Figure 1). The cores provide support for the main research projects, as well as projects from investigators supported by our Developmental Programs, to facilitate and expand translational research for the SPORE. Our Developmental Programs (DRP/CEP) recruit and support qualified investigators who will broadly enhance research in NF1-associated tumors nationwide, with an emphasis on investigators from a broad array of demographic groups. Across all three projects, genome-wide analysis of clinical specimens and murine cancers will elucidate molecular mechanisms involved in tumorigenesis and tumor resistance. Achieving the objectives of Projects 1-3 has the potential to inform new therapeutic approaches for the substantial proportion of human cancers characterized by somatic NF1 and RAS mutations. Given the central importance of aberrant Ras/GAP function in human cancer and the emerging role of somatic NF1 mutations in common sporadic malignancies, achieving the goals of this SPORE will broadly advance translational cancer research and stimulate “next generation” treatments.
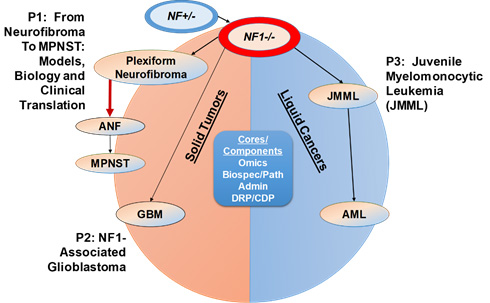
Research components of the DHART SPORE
Project 1: From neurofibroma to MPNST: models, biology and translation to clinic
Project Co-Leaders:
Wade Clapp, MD (IU) (Basic Science Co-Leader)
Jaishri Blakeley, MD (JHU) (Clinical Co-Leader)
Brigitte Widemann, MD (NCI) (Clinical Co-Leader)
Lu Le, MD, PhD (UTSW) (Basic Science Co-Leader)
Aim 1: To implement better combination treatments for Plexiform neurofibromas (PNF), we will test the hypothesis that germline NF1 mutations modulate the biologic properties of sub-populations of NF1+/- fibroblasts within the microenvironment and characterize the ECM signaling factors that interact with the neoplastic Schwann cells to accelerate PNF formation and maintenance. Identifying these components will facilitate new therapeutic strategies for PNF that simultaneously target both tumorigenic Schwann cells and the microenvironment.
Aim 2: We will utilize the novel murine models to extend the preclinical studies performed during our first funding cycle to investigate potential synergism of MEKi combined with the multi-RTK inhibitor cabozantinib or a BETi for treating existing PN and ANP as well as preventing transformation to MPNST.
Aim 3: Parallel studies in PDX models will be conducted. Finally, we will translate our findings in Aim 4 to conduct early phase clinical trials of the most promising agents (including MEKi and cabozantinib combination therapy) with correlative molecular analyses.
The overall translational impact includes:
(1) investigating a rational approach to combinatorial therapy based on simultaneously interfering with paracrine growth signals emanating from the tumor microenvironment while also targeting aberrant Ras/Raf/MEK/ERK pathway activation in tumor cells to treat PNF;
(2) defining novel biomarkers in NF1 patients enrolled on combination trials by analyzing tumor specimens and blood obtained both prior to and after treatment; and,
(3) identifying rational drug combinations in preclinical models that will allow treatment and/or prevention of ANF and MPNSTs that will lead to new phase I-II trials for these tumors in year 2-3 of the proposed funding period.
Project 2: NF1-associated glioblastoma
Project Co-Leaders:
Luis Parada, PhD (MSKCC) (Basic Science Co-Leader)
Ingo Mellinghoff, MD (MSKCC) (Clinical Co-Leader)
To understand how the absence of a functional NF1 gene (somatic or germline) specifically and uniquely affects glioma development and maintenance, we are generating data in murine models, patient-derived models, and primary human tumor samples.
Aim 1: Will focus on experiments in three different GEM models that develop GBM at high penetrance and reflect the three tumor genotypes (i.e., germline NF1 mutant GBM, somatic NF1 mutant GBM and NF1 wild-type GBM). We will use state-of-the-art GEM models ideal for the generation and initial testing of genotype-specific hypothesis to perform in vitro and in vivo comparative molecular and phenotypic studies of NF1 germline or somatic GBM versus non-NF1 mutant GBM. We will extend to analysis of the TME, including microglia and specific immune cell populations across these genotyped tumors.
Aim 2: Will extend studies to a panel of PDX GBM models derived at MSKCC which more closely represent the genetic complexity of human GBM. Proof of principle studies have shown that key phenotypic and molecular features of our GEM models are maintained in the PDX models, providing a template for data integration.
Aim 3: Will focus on the in-depth phenotypic and molecular characterization of human germline NF1 GBM, using multi-site collaboration to enable sufficient patient accrual for this ultra-rare disease and state-of-the art technology for in-situ characterization of the TME to allow cross-analysis between GEM, PDX and human samples.
The overall translational impact includes an in-depth characterization of GBM pathogenesis and investigating how NF1 mutations influence the underlying biology and therapeutic responses of sporadic and germline NF1-mutant GBM.
Project 3: A high content clinical trial of the MEK inhibitor Trametinib in JMML
Project Co-Leaders:
Kevin Shannon, MD (UCSF) (Basic Science Co-Leader)
Elliot Stieglitz, MD (UCSF) (Clinical Co-Leader)
The overall goal is to implement more effective and less toxic treatments for JMML, an aggressive cancer that predominantly affects children <2 years old. Translational studies are of broad interest as JMML is the best example of a human cancer that is initiated by — and dependent on — aberrant Ras/MAPK signaling. Understanding how JMML patients respond to targeted inhibitors is of exceptional interest to investigators working on mechanism- based therapies to treat AML and other more common cancers that acquire somatic NF1, NRAS, and/or KRAS mutations.
Our ongoing national trial (ADVL1521, NCT03190915) of trametinib, an FDA-approved MEK inhibitor, for patients with relapsed/refractory JMML is a key component of this SPORE.
Aim 1: Drs. Loh and Stieglitz will complete the ongoing ADVL1521 trial and will open the first risk-stratified, multi-center interventional study in newly diagnosed JMML patients based on the results of molecular testing and epigenetic signatures. This trial will generate foundational data for a next generation, CLIA-compliant sequencing assay for risk stratification.
Aim 2: We will perform biologic and preclinical studies in GEM models of JMML and acute myeloid leukemia (AML) in order to accelerate clinical translation by testing promising new drugs and drug combinations.
The translational impact of the studies described in this project include:
(1) testing signal transduction inhibition as an alternative therapeutic strategy to HSCT in JMML;
(2) developing a new therapeutic paradigm for JMML based on stratifying patients according to the underlying molecular landscape of their disease and administering mechanism-based personalized therapies that are more effective and less toxic; and,
(3) generating data in GEM models to identify the most promising new drugs and drug combinations that will be evaluated in “next generation” early phase clinical trials in hematologic and non-hematologic cancers characterized by germline and somatic NF1 mutations.
Administrative Core
Core Directors:
Wade Clapp, MD (IU)
Kevin Shannon, MD (UCSF)
The goal of the Administrative Core is to provide scientific and administrative oversight for all the program components of the DHART SPORE. This core serves as the central point for all administrative activities and essential services, including being the organizing unit for the coordination, integration and communication for all components. These coordinating components include:
(1) providing the overall organizational framework by which researchers communicate and interact/collaborate,
(2) managing finances,
(3) supporting the coordination of the review of progress of the research projects, scientific cores and developmental programs (DRP and CEP),
(4) consulting with the members of the External and Internal Advisory boards,
(5) coordinating the overall data integration and analysis of the DHART SPORE, and
(6) continuing to establish and refine the infrastructure to support the meaningful sharing of data and information generated through our studies with other internal and external NF-collaborative consortiums.
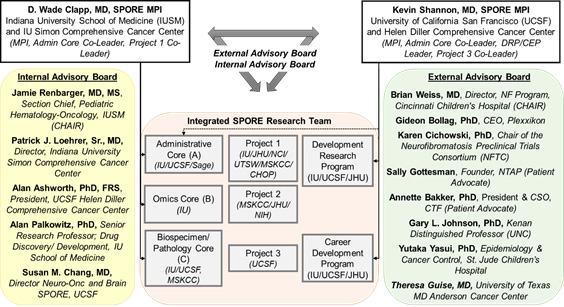
DHART SPORE organization.
Omics Core
Core Director:
Yunlong Liu, PhD (IU)
The Omics Core will advance the scientific goals of Projects 1-3 by providing a unique set of proteomic and genomic platforms for performing RNAseq and whole exome sequencing analysis combined with comprehensive kinome activation measurements. The overall goal of this core is to provide state-of-the-art support for research design consultation, performance of genomics and kinomics experiments, and integrated data analysis to DHART investigators to elucidate changes during NF1-related tumor development and in response to therapy. The application of -omics approaches facilitates the interrogation of research hypotheses while providing a snapshot of other pathways that could be intimately involved in previously unappreciated ways. Considering the high number of connections between the DHART researchers, -omics experiments will both strengthen and expand these efforts, leading to an increased understanding of the biology of NF1-associated cancers and identification of new targeted molecular therapies. Core B will continue to focus on the genome, transcriptome, and kinome as well as their integration through interactome and signaling pathway analysis. The DHART projects will benefit from the ongoing application/development of omics approaches at IUSM including next generation sequencing, single cell analytics and kinomic profiling capacity. As illustrated in the figure below, the Omics Core represents an alliance of existing genomics, proteomics and kinomics, and bioinformatics centers that have been established at IUSM, and brings together a wealth of expertise in systems-level analysis of cells and tissues for interrogation of biological perturbation.
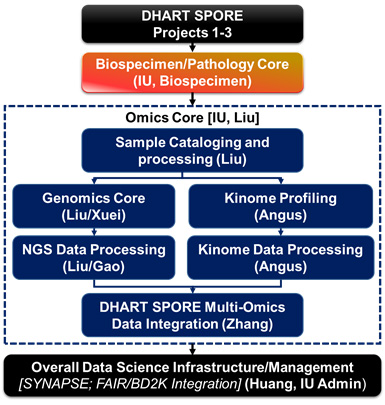
Overview of Omics Core Process.
Biospecimen and Pathology Core
Core Directors:
Tatiana Foroud, PhD (IU)
Scott Kogan, MD (USCF)
Melike Pekmezci, MD (UCSF)
Tejus A. Bale, MD (MSKCC)
The DHART Biospecimen and Pathology Core brings together a team with expertise in all relevant subspecialties of pathologic analyses and diagnostics as well as in the collecting, storing and sharing blood and tissue samples to advance the scientific goals of the DHART SPORE. Specifically, investigators in this core lead programs related to biospecimen banking, genomics, molecular pathogenesis of sarcomas, peripheral and central nervous system tumors and leukemogenesis. This core group plays an integral role within this SPORE by: (1) tracking all human and mouse samples from the projects (and any Developmental Program awards) in the SPORE; (2) performing pathological review of samples as requested by investigators, (3) independent validation of specimen diagnosis of tissues; (4) making histologic and molecular data available via a shared platform and (5) linking tumor specimens to radiographic images and de-identified data.
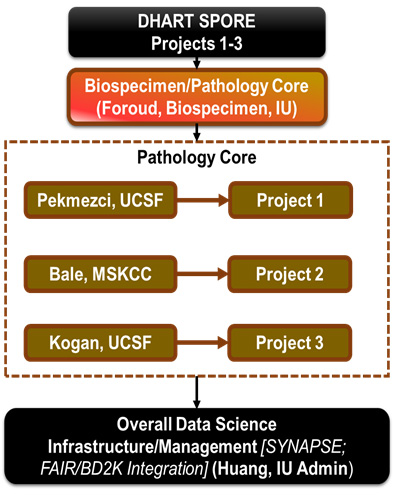
Overview of Biospecimen and Pathology Core.
Developmental Research Program
Program Directors:
Kevin Shannon, MD (UCSF)
Jaishri Blakeley, MD (JHU)
The DHART SPORE Developmental Research Program (DRP) will support innovative pilot projects to advance the diagnosis and treatment of early stage tumors and advanced cancers characterized by germline and somatic Neurofibromatosis type 1 (NF1) mutations. The DRP is designed to recruit and support established investigators to develop a new research theme in the field of NF1-related cancers. We highly encourage investigators from a broad array of demographic groups to apply for DRP projects.
Career Enhancement Program
Program Directors:
Kevin Shannon, MD (UCSF)
Jaishri Blakeley, MD (JHU)
The DHART SPORE Career Enhancement Program (CEP) is designed to solicit, support, and advance the careers of junior researchers, at the faculty Instructor and Assistant Professor levels, to engage in translational research focused on NF1/Rasopathy disorders and cancer. We encourage qualified scientists from a broad array of demographic groups to apply.